|
En química la Dinámica Molecular (DM) es una técnica de simulación en la que se permite que átomos y moléculas interactúen por un periodo de tiempo. En general, los sistemas moleculares son complejos y consisten de un gran número de partículas, por lo cual sería imposible encontrar sus propiedades de forma analítica. Para evitar este problema, la DM utiliza métodos numéricos. Representa un punto intermedio entre los experimentos y la teoría. Puede ser entendida como un experimento en la computadora. La dinámica molecular es un campo interdisciplinario. Sus leyes y teorías provienen de las Matemáticas, Física y Química. Emplea algoritmos de las Ciencias de la Computación y Teoría de la información. Permite entender a los materiales y las moléculas no cómo entidades rígidas, sino como cuerpos animados. Originalmente fue concebida dentro de la física teórica, aunque hoy en día se le utiliza sobre todo en biofísica y ciencia de materiales. Su campo de aplicación va desde superficies catalíticas hasta sistemas biológicos. Esta técnica presenta un compromiso entre costo computacional y fiabilidad en los resultados, ya que se utilizan las Ecuaciones de Newton, que son menos costosas que las de la mecánica cuántica. Es por ello que muchas propiedades que pueden resultar de interés, como por ejemplo la formación o ruptura de enlaces no puedan ser estudiadas mediante este método ya que no contempla estados excitados o reactividad. Existen métodos híbridos denominados QM/MM (Quantum Mechanics/Molecular Mechanics) en los que un centro reactivo es tratado de modo cuántico mientras que el ambiente que lo rodea se trata de modo clásico. El desafío en este tipo de métodos resulta en la definición de manera precisa de la interacción entre los dos formas de describir el sistema. Sabemos que la materia está constituída de partículas en movimiento e interacción al menos desde la época de Boltzmann en el siglo 19. Pero muchos aún se imaginan a las moléculas como los modelos estáticos de un museo. Richard Feynman dijo en 1963 que "todo lo que hacen los seres vivos puede ser entendido a través de los saltos y contorsiones de los átomos."[1] Una de las contribuciones más importantes de la dinámica molecular es crear conciencia de que el DNA y las proteínas son máquinas en movimiento. Se le utiliza para explorar la relación entre estructura, movimiento y función. También se le ha llamado "estadística mecánica numérica" o "la visión de Laplace de la mecánica Newtoniana", en el sentido de predecir el futuro al animar las fuerzas de la naturaleza. Resulta tentador pensar que la DM es como un microscopio virtual. Sin embargo, las simulaciones largas están mal condicionadas, lo cual genera errores numéricos acumulativos. Esto quiere decir que debemos abandonar la ilusión de que estamos siguiendo el comportamiento real de una molécula en el tiempo. De cualquier forma, la dinámica molecular nos permite explorar su comportamiento representativo en el espacio fase.
El resultado de una simulación de dinámica molecular son las posiciones X y velocidades V de cada átomo de la molécula, para cada instante en el tiempo. A esto se le llama trayectoria. Conocimientos adicionales recomendados
Principios físicosConjunto microcanónico (NVE)La forma más simple de dinámica molecular ocurre en el conjunto microcanónico. En él, el sistema está aislado: su volumen no se altera (V) y no intercambia masa (N) ni energía (E) con el entorno. Para un sistema de N partículas con coordenadas X y velocidades V, se puede plantear el siguiente par de ecuaciones diferenciales de primer orden La función de energía potencial U(X) son las atracciones y repulsiones que sienten los átomos entre sí debido a los enlaces químicos, interacciones electrostáticas, van der Waals, etc. de las moléculas. A U(X) también se le conoce como campo de fuerza y es una función de las coordenadas de las partículas X. Normalmente proviene de cálculos de química cuántica y/o experimentos espectroscópicos. Sin embargo, el campo de fuerza generalmente tiene una forma funcional que lo hace pertenecer a la mecánica clásica. La trayectoria de las partículas es discreta en el tiempo. Normalmente se elige un paso de tiempo suficientemente pequeño (p.ej. 1 femtosegundo) para evitar errores numéricos de discretización. Para cada paso de tiempo, se integra la posición X y velocidad V con un método simpléctico como la integración de Verlet. Dadas las posiciones iniciales (p.ej. la estructura de rayos X de una proteína) y las velocidades iniciales (p.ej. aleatorias y Gaussianas), es posible calcular todas las posiciones y velocidades en el futuro. SoftwareEn la actualidad existen varios programas capaces de llevar a cabo las simulaciones de DM, y a su vez existen varios campos de fuerza, que en general pueden utilizarse con diversos programas. AMBER, NAMD y GROMACS son algunos de los paquetes más populares. Referencias
Véase tambiénEnlaces externos
Categoría: Química |
|
| Este articulo se basa en el articulo Dinámica_molecular publicado en la enciclopedia libre de Wikipedia. El contenido está disponible bajo los términos de la Licencia de GNU Free Documentation License. Véase también en Wikipedia para obtener una lista de autores. |



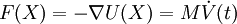
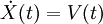

Acerca de quimica.es
Descubra toda la información interesante sobre nuestro portal especializado quimica.es.
Acerca de LUMITOS
Descubra más información sobre la empresa LUMITOS y nuestro equipo.
Anunciarse en LUMITOS
Descubra cómo puede ayudarle LUMITOS en su marketing online.
Los portales especializados de LUMITOS







© 1997-2024 LUMITOS AG, All rights reserved

Enciclopedia