|
La cinética enzimática estudia la velocidad de las reacciones químicas que son catalizadas por las enzimas. El estudio de la cinética de una enzima permite explicar los detalles de su mecanismo catalítico, su papel en el metabolismo, cómo es controlada su actividad en la célula y cómo puede ser inhibida su actividad por fármacos o venenos o potenciada por otro tipo de moléculas. Las enzimas son proteínas (macromoléculas) con la capacidad de manipular otras moléculas, denominadas sustratos. Un sustrato es capaz de unirse al centro catalítico de la enzima que lo reconozca y transformarse en un producto a lo largo de una serie de pasos denominados mecanismo enzimático. Algunas enzimas pueden unir varios sustratos diferentes y/o liberar diversos productos, como es el caso de las proteasas al romper una proteína en dos polipéptidos. En otros casos, se produce la unión simultánea de dos sustratos, como en el caso de la ADN polimerasa, que es capaz de incorporar un nucleótido (sustrato 1) a una hebra de ADN (sustrato 2). Aunque todos estos mecanismos suelen seguir una compleja serie de pasos, también suelen presentar una etapa limitante que determina la velocidad final de toda la reacción. Esta etapa limitante puede consistir en una reacción química o en un cambio conformacional de la enzima o del sustrato. El conocimiento adquirido acerca de la estructura de las enzimas ha sido de gran ayuda en la visualización e interpretación de los datos cinéticos. Por ejemplo, la estructura puede sugerir cómo permanecen unidos sustrato y producto durante la catálisis, qué cambios conformacionales ocurren durante la reacción, o incluso el papel en particular de determinados residuos aminoácidos en el mecanismo catalítico. Algunas enzimas modifican su conformación significativamente durante la reacción, en cuyo caso, puede ser crucial saber la estructura molecular de la enzima con y sin sustrato unido (se suelen usar análogos que se unen pero no permiten llevar a cabo la reacción y mantienen a la enzima permanentemente en la conformación de sustrato unido). Los mecanismos enzimáticos pueden ser divididos en mecanismo de único sustrato o mecanismo de múltiples sustratos. Los estudios cinéticos llevados a cabo en enzimas que solo unen un sustrato, como la triosafosfato isomerasa, pretenden medir la afinidad con la que se une el sustrato y la velocidad con la que lo transforma en producto. Por otro lado, al estudiar una enzima que une varios sustratos, como la dihidrofolato reductasa, la cinética enzimática puede mostrar también el orden en el que se unen los sustratos y el orden en el que los productos son liberados. Sin embargo, no todas las catálisis biológicas son llevadas a cabo por enzimas proteicas. Existen moléculas catalíticas basadas en el ARN, como las ribozimas y los ribosomas, esenciales para el splicing alternativo y la traducción del ARNm, respectivamente. La principal diferencia entre las ribozimas y las enzimas radica en el limitado número de reacciones que pueden llevar a cabo las primeras, aunque sus mecanismos de reacción y sus cinéticas pueden ser estudiadas y clasificadas por los mismos métodos. Conocimientos adicionales recomendados
Principios generalesLa reacción catalizada por una enzima utiliza la misma cantidad de sustrato y genera la misma cantidad de producto que una reacción no catalizada. Al igual que ocurre en otros tipos de catálisis, las enzimas no alteran en absoluto el equilibrio de la reacción entre sustrato y producto.[1] Sin embargo, al contrario que las reacciones químicas, las enzimas se saturan. Esto significa que a mayor cantidad de sustrato, mayor número de centros catalíticos estarán ocupados, lo que incrementará la eficiencia de la reacción, hasta el momento en que todos los sitios posibles estén ocupados. En ese momento se habrá alcanzado el punto de saturación de la enzima y, aunque se añada más sustrato, no aumentará más la eficiencia. Las dos propiedades cinéticas más importantes de una enzima son: el tiempo que tarda en saturarse con un sustrato en particular y la máxima velocidad de reacción que pueda alcanzar. El conocimiento de estas propiedades hace posible hipotetizar acerca del comportamiento de una enzima en el ambiente celular y predecir cómo responderá frente a un cambio de esas condiciones. Ensayos enzimáticosUn ensayo enzimático es un procedimiento, llevado a cabo en un laboratorio, mediante el cual se puede medir la velocidad de una reacción enzimática. Como las enzimas no se consumen en la reacción que catalizan, los ensayos enzimáticos suelen medir los cambios experimentados bien en la concentración de sustrato (que va decreciendo), bien en la concentración de producto (que va aumentando). Existen diversos métodos para realizar estas medidas. La espectrofotometría permite detectar cambios en la absorbancia de luz por parte del sustrato o del producto (según la concentración de estos) y la radiometría implica incorporación o liberación de radiactividad para medir la cantidad de producto obtenido por tiempo. Los ensayos espectrofotométricos son los más utilizados, ya que permiten medir la velocidad de la reacción de forma continua. Por el contrario, los ensayos radiométricos requieren retirar las muestras para medirlas, por lo que son ensayos discontinuos. Sin embargo, estos ensayos son extremadamente sensibles y permiten detectar niveles muy bajos de actividad enzimática.[2] También se puede utilizar la espectrometría de masas para detectar la incorporación o liberación de isótopos estables cuando el sustrato es convertido en producto. Los ensayos enzimáticos más sensibles utilizan láseres dirigidos a través de un microscopio para observar los cambios producidos en enzimas individuales cuando catalizan una reacción. Estas medidas pueden utilizar cambios producidos en la fluorescencia de cofactores que intervienen en el mecanismo de catálisis o bien unir moléculas fluorescentes en lugares específicos de la enzima, que permitan detectar movimientos ocurridos durante la catálisis.[3] Estos estudios están dando una nueva visión de la cinética y la dinámica de las moléculas individuales, en oposición a los estudios de cinética enzimática tradicionales, en los que se observa y se mide el comportamiento de una población de millones de moléculas de enzima. En la figura de la derecha se puede observar la típica evolución de una curva obtenida en un ensayo enzimático. Inicialmente, la enzima transforma el sustrato en producto siguiendo un comportamiento lineal. A medida que avanza la reacción, se va agotando la cantidad de sustrato y va disminuyendo la cantidad de producto que se genera por unidad de tiempo (disminuye la velocidad de la reacción), lo que se manifiesta en forma de curva asintótica en la gráfica. Dependiendo de las condiciones del ensayo y del tipo de enzima, el período inicial puede durar desde milisegundos hasta horas. Los ensayos enzimáticos suelen estar estandarizados para que el período inicial dure en torno a un minuto, para llevar a cabo las medidas más fácilmente. Sin embargo, los modernos equipos de mezcla rápida de líquidos permiten llevar a cabo medidas cinéticas de períodos iniciales cuya duración puede llegar a ser inferior a un segundo.[4] Este tipo de ensayos rápidos son esenciales para medidas de la cinética del estado estacionario, discutida más abajo. La mayoría de los estudios de cinética enzimática se centran en el período inicial, es decir, en la zona lineal de la reacción enzimática. Sin embargo, también es posible medir toda la curva de la reacción y ajustar estos datos a una ecuación no lineal. Esta forma de medir las reacciones enzimáticas es denominada análisis de la curva de progreso.[5] Esta aproximación es muy útil como alternativa a las cinéticas rápidas, cuando el período inicial es demasiado rápido para ser medido con precisión. Factores físico-químicos que pueden modificar la actividad enzimática
Reacciones con un sustratoLas enzimas que presentan un mecanismo de único sustrato incluyen isomerasas, tales como la triosafosfato isomerasa o la bisfosfoglicerato mutasa, y liasas intramoleculares, tales como la adenilato ciclasa o la ribozima ARN-liasa.[6] Sin embargo, existen ciertas reacciones enzimáticas de único sustrato que no pertenecen a esta categoría de mecanismos, como es el caso de la reacción catalizada por la catalasa. La catalasa reacciona inicialmente con una molécula de peróxido de hidrógeno (agua oxigenada) y queda en un estado oxidado tras liberar el producto (agua), y, posteriormente, es reducida por una segunda molécula de sustrato. Aunque durante la reacción solo participa un sustrato, la existencia de un intermediario enzimático modificado permite incluir al mecanismo de la catalasa en la categoría de mecanismos de ping-pong, un tipo de mecanismo discutido más adelante. Cinética de Michaelis-MentenComo las reacciones catalizadas por enzimas son saturables, la velocidad de catálisis no muestra un comportamiento lineal en una gráfica al aumentar la concentración de sustrato. Si la velocidad inicial de la reacción se mide a una determinada concentración de sustrato (representado como [S]), la velocidad de la reacción (representado como V) aumenta linealmente con el aumento de la [S], como se puede ver en la figura. Sin embargo, cuando aumentamos la [S], la enzima se satura de sustrato y alcanza su velocidad máxima (Vmax), que no sobrepasará en ningún caso, independientemente de la [S].
El modelo de cinética michaeliana para una reacción de único sustrato se puede ver en la figura de la izquierda. Primeramente, tiene lugar una reacción química bimolecular entre la enzima E y el sustrato S, formándose el complejo enzima-sustrato ES. Aunque el mecanismo enzimático para una reacción unimolecular
k2 también llamado kcat o número de recambio, hace referencia al máximo número de reacciones enzimáticas catalizadas por segundo.
La ecuación de Michaelis-Menten[7] describe como la velocidad de la reacción depende del equilibrio entre la [S] y la constante k2. Leonor Michaelis y Maud Menten demostraron que si k2 es mucho menor que k1 (aproximación del equilibrio) se puede deducir la siguiente ecuación:
Esta famosa ecuación es la base de la mayoría de las cinéticas enzimáticas de sustrato único. La constante de Michaelis Km se define como la concentración a la que la velocidad de la reacción enzimática es la mitad de la Vmax. Esto puede verificarse sustituyendo la concentración de sustrato por dicha constante ([S] = Km). Si la etapa limitante de la velocidad de la reacción es lenta comparada con la disociación de sustrato (k2 <<< k1), la constante de Michaelis Km será aproximadamente la constante de disociación del complejo ES, aunque sea una situación relativamente rara. La situación más común, donde k2 > k1, es denominada cinética de Briggs-Haldane.[8] La ecuación de Michaelis-Menten aún se mantiene bajo estas condiciones más generales, como puede derivarse de la aproximación del estado estacionario. Durante el período inicial, la velocidad de la reacción es más o menos constante, indicando que la [ES] también se mantendrá constante: De esta forma, la concentración de ES viene dada por la siguiente expresión: donde la constante de Michaelis Km se define así: Con lo cual, después de operar todos los factores, obtenemos una fórmula general para la velocidad de la reacción que coincide con la ecuación de Michaelis-Menten: La constante de especificidad kcat / Km mide la eficiencia con la que una enzima convierte un sustrato en producto. Utilizando la definición de la constante de Michaelis Km, la ecuación de Michaelis-Menten podría escribirse de la siguiente forma: donde [E] es la concentración de enzima libre. Así, la constante de especificidad se convierte en una constante bimolecular efectiva de la enzima libre que reacciona con sustrato libre para formar producto. Esta constante viene definida por la frecuencia con la que el sustrato y la enzima se encuentran en una solución, y ronda aproximadamente un valor de 1010 M-1 s-1 a 25º C. Curiosamente, este máximo no depende del tamaño del sustrato o de la enzima. La proporción de las constantes de especificidad para dos sustratos es una comparación cuantitativa de la eficiencia de la enzima para convertir en productos dichos sustratos. La pendiente de la ecuación de Michaelis-Menten a bajas concentraciones de sustrato (cuando [S] << Km) también proporciona la constante de especificidad. Representación de la ecuación de Michaelis-MentenLa gráfica de velocidad frente a [S] mostrada anteriormente no es lineal. Aunque a bajas concentraciones de sustrato se mantenga lineal, se va curvando a medida que aumenta la concentración de sustrato. Antes de la llegada de los ordenadores, que permiten ajustar regresiones no lineales de forma sencilla, podía llegar a ser realmente difícil estimar los valores de la Km y la Vmax en las gráficas no lineales. Esto dio lugar a que varios investigadores concentraran sus esfuerzos en desarrollar linearizaciones de la ecuación de Michaelis-Menten, dando como resultado la gráfica de Lineweaver-Burke y el diagrama de Eadie-Hofstee. Con el siguiente tutorial de la cinética de Michaelis-Menten realizado en la Universidad de Virginia α, se puede simular el comportamiento de una enzima variando las constantes cinéticas. La gráfica de Lineweaver-Burk o representación de doble recíproco es la forma más común de mostrar los datos cinéticos. Para ello, se toman los valores inversos a ambos lados de la ecuación de Michaelis-Menten. Como se puede apreciar en la figura de la derecha, el resultado de este tipo de representación es una línea recta cuya ecuación es y = mx + c, siendo el punto de corte entre la recta y el eje de ordenadas equivalente a 1/Vmax, y el punto de corte entre la recta y el eje de abscisas equivalente a -1/Km.
Significado de las constantes cinéticasLa importancia del estudio de la cinética enzimática reside en dos principios básicos. En primer lugar, permite explicar cómo funciona una enzima, y en segundo lugar, permite predecir cómo se comportará esa enzima in vivo. Las constantes cinéticas definidas anteriormente, Km y Vmax, son los pilares fundamentales a la hora de intentar comprender el funcionamiento de las enzimas en el control del metabolismo. Sin embargo, llevar a cabo estas predicciones no es trivial, incluso en los sistemas más simples. Por ejemplo, la malato deshidrogenasa sintetiza, en el interior de la mitocondria, oxalacetato, el cual puede ser sustrato de diversas enzimas como la citrato sintasa, en el ciclo de los ácidos tricarboxílicos, la fosfoenolpiruvato carboxiquinasa, en la gluconeogénesis, o la aspartato aminotransferasa, en la biosíntesis de ácido aspártico. Para ser capaz de predecir cuánto oxalacetato será desviado por cada una de las rutas es necesario saber tanto la concentración del oxalacetato como la concentración y los parámetros cinéticos de cada una de las enzimas. Este ejemplo denota la complejidad que podemos llegar a encontrar al intentar predecir el comportamiento de rutas metabólicas completas o de organismos enteros, por medio de modelos matemáticos. Aunque estos objetivos aún no se han alcanzado en eucariotas, se han obtenido ciertos progresos en bacterias, utilizando modelos del metabolismo de Escherichia coli.[11] [12] Reacciones multisustratoLas reacciones multisustrato siguen una serie de complejas ecuaciones que describen cómo se unen los sustratos y en qué orden lo hacen. El análisis de estas reacciones es mucho más sencillo si la concentración del sustrato A se mantiene constante y la del sustrato B varía. En estas condiciones, la enzima se comporta igual que una enzima de único sustrato, por lo que en una gráfica de velocidad la concentración de sustrato dará unos valores aparentes de las constantes cinéticas, Km y Vmax, para el sustrato B. Si se realizan una serie de medidas a diferentes concentraciones fijas de sustrato A, los datos obtenidos permitirán saber a qué tipo de mecanismo pertenece la reacción enzimática. Para una enzima que una dos sustratos A y B, y los transforme en dos productos P y Q, existen dos tipos de mecanismos descritos hasta ahora. Mecanismo de complejo ternarioLas enzimas (E) que presentan este mecanismo de reacción unen al mismo tiempo los dos sustratos (A y B), dando lugar a un complejo ternario EAB. El orden secuencial de unión de los sustratos puede ser al azar (mecanismo al azar) o seguir un orden en particular (mecanismo ordenado). Si fijamos la concentración del sustrato A y variamos la de B, y representamos gráficamente el comportamiento de la enzima mediante un diagrama de Lineweaver-Burke, obtendremos una serie de rectas con un punto de intersección común a todas ellas. Entre las enzimas que presentan este mecanismo podemos encontrar la glutation S-transferasa,[13] la dihidrofolato reductasa[14] y la ADN polimerasa.[15] Los siguientes enlaces muestran animaciones del mecanismo de complejo ternario de la dihidrofolato reductasa β y de la ADN polimerasa γ. Mecanismo de ping-pongComo se puede apreciar en la figura de la derecha, las enzimas con un mecanismo de ping-pong pueden presentar dos estados, la conformación normal (E) y la conformación modificada químicamente (E*) o conformación intermedia. En este tipo de mecanismo, el sustrato A se une a la enzima E, que pasa a un estado intermedio E*, por ejemplo, por transferencia de un grupo químico al centro activo de la enzima, pudiendo ya ser liberado en forma de producto P. Únicamente cuando el sustrato A ya ha sido liberado del centro activo de la enzima puede unirse el sustrato B, que devuelve a la enzima modificada E* a su estado original E, y liberarlo en forma de producto Q. Si fijamos la concentración de A y variamos la de B, y representamos gráficamente una enzima con mecanismo de ping-pong en un diagrama de Lineweaver-Burke, obtendremos una serie de rectas paralelas entre sí. Entre las enzimas con este tipo de mecanismo podemos encontrar alguna oxidorreductasa, como la tiorredoxima peroxidasa,[16] transfereasas, como la acil-neuraminato citidil transferasa,[17] y serin proteasas, como la tripsina y la quimiotripsina.[18] Las serin-proteasas conforman una diversa familia de enzimas muy comunes, que incluyen enzimas digestivas (tripsina, quimiotripsina y elastasa), varias enzimas del proceso de coagulación y muchas otras. En las serin-proteasas, el estado intermedio E* es una especie acilada en una serina del centro catalítico de la enzima. El siguiente enlace muestra una animación del mecanismo catalítico de la quimiotripsina δ. Cinéticas no MichaelianasAlgunas reacciones enzimáticas dan lugar a curvas sigmoideas, al ser representadas en una curva de saturación, lo que suele indicar una unión cooperativa del sustrato al centro catalítico de la enzima. Esto quiere decir que la unión de una molécula de sustrato influye en la unión de las moléculas de sustrato posteriores. Este comportamiento es el más común en las enzimas multiméricas, que presentan varias zonas de interacción con el sustrato.[19] El mecanismo de cooperación es semejante al observado en la hemoglobina. La unión de una molécula de sustrato a una de las zonas de interacción altera significativamente la afinidad por el sustrato de las demás zonas de interacción. Las enzimas con este tipo de comportamiento son denominadas alostéricas. La cooperatividad positiva tiene lugar cuando la primera molécula de sustrato unida incrementa la afinidad del resto de zonas de interacción. Por el contrario, la cooperatividad negativa tiene lugar cuando la primera molécula de sustrato unida reduce la afinidad de la enzima por nuevas moléculas de sustrato. Como ejemplos de enzimas con cooperatividad positiva tenemos la aspartato transcarbamilasa bacteriana[20] y la fosfofructoquinasa,[21] y con cooperatividad negativa, la tirosil ARNt-transferasa de mamíferos.[22] La cooperatividad es un fenómeno bastante común y puede llegar a ser crucial en la regulación de la respuesta enzimática a cambios en la concentración de sustrato. La cooperatividad positiva hace que la enzima sea mucho más sensible a la concentración de sustrato, con lo que su actividad puede llegar a variar en gran medida aunque se mueva en rangos muy estrechos de concentración de sustrato. Por el contrario, la cooperatividad negativa hace que la enzima sea insensible a pequeños cambios en la concentración de sustrato. La ecuación de Hill[23] suele ser utilizada para describir cuantitativamente el grado de cooperatividad en cinéticas no michaelianas. El coeficiente de Hill (n) indica cuántas de las zonas de unión de sustrato de una enzima afectan a la afinidad de la unión del sustrato en el resto de las zonas de unión. El coeficiente de Hill puede tomar valores mayores o menores que 1:
Cinética del estado pre-estacionarioAl realizar un ensayo enzimático y mezclar la enzima con el sustrato, existe un breve período inicial en el que no se produce ni síntesis de producto, ni estado intermedio de la enzima. El estudio de los milisegundos siguientes a la mezcla es denominado cinética del estado pre-estacionario y está relacionado con la formación y consumo de los intermediarios enzima-sustrato (ES o E*) hasta el momento en el que se alcanzan ciertas concentraciones y comienza el estado estacionario. La primera enzima en la que se estudió este proceso, durante la reacción de hidrólisis, fue la quimiotripsina.[24] La detección de intermediarios suele ser la principal evidencia necesaria para saber bajo qué mecanismo actúa la enzima. Por ejemplo, al realizar un ensayo de cinética rápida de una reacción enzimática gobernada por un mecanismo de ping-pong, podremos hacer un seguimiento de la liberación de producto P y medir la formación de intermediarios enzimáticos modificados E*.[25] En el caso de la quimiotripsina, el intermediario enzimático se produce por un ataque nucleofílico del sustrato sobre una serina del centro catalítico, lo que genera una forma intermediaria acilada de la quimiotripsina. En la figura de la derecha, se puede observar como la enzima pasa rápidamente a un estado intermedio E* durante los primeros segundos de la reacción. Posteriormente, cuando es alcanzado el estado estacionario, la velocidad disminuye. Esta primera fase de la reacción proporciona la tasa de conversión de la enzima. Por lo tanto, la cantidad de producto liberado durante esta fase (que puede obtenerse prolongando la recta correspondiente al estado estacionario hasta cortar el eje y) también da la cantidad de enzima funcional presente en el ensayo.[26] Mecanismo químicoUno de los objetivos más importantes en los estudios de la cinética enzimática es determinar el mecanismo químico que subyace en la reacción enzimática, por ejemplo, determinar la secuencia ordenada de sucesos que transcurren en la transformación de sustrato en producto. Las aproximaciones cinéticas discutidas anteriormente pueden proporcionar información relacionada con la velocidad de reacción de los intermediarios enzimáticos formados, pero no permitirán identificar qué intermediarios son exactamente. Con el fin de determinar la etapa limitante de la reacción o los intermediarios que se forman durante la misma, se pueden llevar a cabo ensayos enzimáticos en diversas condiciones con enzimas o sustratos ligeramente modificados, que aporten datos al respecto. Un ejemplo característico de etapa limitante de una reacción es aquella en la que se rompe un enlace covalente dando lugar a un átomo de hidrógeno. Si intercambiamos cada átomo de hidrógeno por su isótopo estable, deuterio, podremos saber cual de los posibles hidrógenos transferidos es el que determina la etapa limitante. La velocidad sufrirá una variación cuando el hidrógeno crítico sea reemplazado, debido al efecto isotópico cinético primario, cuyo origen se encuentra en la mayor estabilidad de los puentes de deuterio, más difíciles de romper que los puentes de hidrógeno.[27] También es posible medir efectos similares por medio de otras sustituciones isotópicas, tales como 13C/12C ó 18O/16O, pero los efectos producidos en estos casos son más difíciles de detectar. Los isótopos también pueden ser utilizados para obtener información acerca del destino de diversas partes de la molécula de sustrato cuando este es transformado en producto. Por ejemplo, a veces es difícil de determinar el origen de un átomo de oxígeno en el producto final, ya que puede provenir del agua o de la molécula de sustrato. Esto puede resolverse mediante la sustitución sistemática de los átomos de oxígeno de las moléculas que participen en la reacción, por su isótopo estable 18O, llevando posteriormente a cabo una búsqueda del isótopo en el producto obtenido. El mecanismo químico también puede ser elucidado estudiando los efectos cinéticos e isotópicos bajo diferentes condiciones de pH,[28] alterando los niveles de iones metálicos u otros cofactores,[29] por mutagénesis dirigida de aminoácidos conservados, o mediante el estudio del comportamiento de la enzima en presencia de análogos del sustrato. Inhibición enzimáticaLos inhibidores enzimáticos son moléculas que reducen o anulan la actividad enzimática. Estos inhibidores pueden unirse a las enzimas de forma reversible (la desaparición del inhibidor restaura la actividad enzimática) o irreversible (el inhibidor inactiva permanentemente a la enzima). Inhibidores reversiblesLos inhibidores enzimáticos reversibles pueden ser clasificados como competitivos, acompetitivos, no competitivos y mixtos, según el efecto que produzcan en las constantes cinéticas Km y Vmax. Este efecto dependerá de que el inhibidor se una a la enzima E, al complejo enzima-sustrato ES o a ambos, como se puede observar en la figura de la derecha y en la tabla inferior. Para clasificar correctamente un inhibidor pueden llevarse a cabo estudios de su cinética enzimática en función de la concentración de inhibidor. Los cuatro tipos de inhibición dan lugar a diagramas de Lineweaver-Burke y de Eadie-Hofstee[30] que varían de diferente forma con la concentración de inhibidor. Para abreviar, se usan dos símbolos:
donde Ki y K'i son las constantes de disociación para unirse a la enzima y al complejo enzima-sustrato, respectivamente. En presencia de un inhibidor reversible, los valores aparentes de la Km y la Vmax pasan a ser (α/α')Km y (1/α')Vmax, respectivamente, como se puede apreciar en la tabla inferior para los casos más comunes.
Los ajustes por regresión no lineal de los datos de cinética enzimática[31] pueden proporcionar estimaciones muy precisas de las constantes de disociación Ki y Ki'. Inhibidores irreversiblesLos inhibidores enzimáticos también pueden unirse e inactivar una enzima de forma irreversible, generalmente por medio de modificaciones covalentes de residuos del centro catalítico de la enzima. Estas reacciones decaen de forma exponencial y suelen ser saturables. Por debajo de los niveles de saturación, mantienen una cinética de primer orden con respecto al inhibidor. Mecanismos de catálisisEl modelo de ajuste inducido es el más utilizado al realizar estudios de interacción enzima-sustrato.[32] Este modelo propone que las interacciones iniciales entre la enzima y el sustrato son relativamente débiles, pero suficientes para producir ciertos cambios conformacionales en la enzima, que estabilizan e incrementan la fuerza de la interacción. Estos cambios conformacionales implican a una serie de aminoácidos catalíticos del centro activo, en los cuales se producen los enlaces químicos correspondientes entre la enzima y el sustrato. Después de la unión, uno o más mecanismos de catálisis disminuyen la energía del estado de transición de la reacción, por medio de una ruta alternativa a la reacción. Los mecanismos de catálisis se clasifican en base a diferentes criterios: catálisis covalente, catálisis por proximidad y alineación de orbitales, catálisis ácido-base general, catálisis por iones metálicos y catálisis electrostática. Los estudios de cinética enzimática no permiten obtener el tipo de catálisis utilizada por una enzima. Sin embargo, algunos datos cinéticos pueden proporcionar una serie de indicios que lleven a realizar otro tipo de estudios dirigidos a determinar dicho tipo de catálisis. Por ejemplo, en un mecanismo de ping-pong la cinética del estado pre-estacionario puede estar sugiriendo una catálisis de tipo covalente. Por otro lado, variaciones en el pH pueden producir efectos drásticos en la Vmax pero no en la Km, lo que podría indicar que un residuo del centro activo precisa un estado concreto de ionización para que se pueda llevar a cabo la catálisis enzimática. Véase tambiénNotas
Referencias
Para más información
Enlaces externos
|
|||||||||||||||||||||||||
| Este articulo se basa en el articulo Cinética_enzimática publicado en la enciclopedia libre de Wikipedia. El contenido está disponible bajo los términos de la Licencia de GNU Free Documentation License. Véase también en Wikipedia para obtener una lista de autores. |



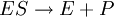 puede ser bastante complejo, existe una etapa enzimática limitante que permite que el mecanismo sea simplificado como una etapa cinética única cuya constante es k2.
puede ser bastante complejo, existe una etapa enzimática limitante que permite que el mecanismo sea simplificado como una etapa cinética única cuya constante es k2.
![\begin{matrix}v = k_2 [\mbox{ES}]\end{matrix}](images/math/e/2/9/e29d34fb0bb15044393f5776866a9eb7.png) (Ecuación 1)
(Ecuación 1)
![[\mbox{E}]_{tot} \ \stackrel{\mathrm{def}}{=}\ [\mbox{E}] + [\mbox{ES}]](images/math/d/4/6/d46887dfbde0b79d341e59f6e9d76fa1.png)
![v = \frac{V_{max}[\mbox{S}]}{K_m + [\mbox{S}]}](images/math/f/1/8/f18456dcd1d181d08cab917a783bd12a.png) (Ecuación 2)
(Ecuación 2)
![\frac{d}{dt}[\mbox{ES}] = k_{1} [\mbox{E}][\mbox{S}] - k_{2}[\mbox{ES}] - k_{-1}[\mbox{ES}] \approx 0](images/math/3/a/f/3afdbe88760ecf976b2f372b5ef1e642.png)
![[\mbox{ES}] \approx \frac{[\mbox{E}]_{tot} [\mbox{S}]}{[\mbox{S}] + K_{m}}](images/math/3/3/3/3338d61e43da719f84794e7c84590e12.png)
![K_{m} \ \stackrel{\mathrm{def}}{=}\ \frac{k_{2} + k_{-1}}{k_{1}} \approx \frac{[\mbox{E}][\mbox{S}]}{[\mbox{ES}]}](images/math/a/6/7/a671ca20a27ae57450854d233e1329f4.png)
![v = k_{2} [\mathrm{ES}] = \frac{k_{2} [\mbox{E}]_{tot} [\mbox{S}]}{[\mbox{S}] + K_{m}} = \frac{V_{max} [\mbox{S}]}{[\mbox{S}] + K_{m}}](images/math/6/b/a/6ba7f7007a60a9dde4714e06d6d11e62.png)
![v = k_{2} [\mathrm{ES}] = \frac{k_{2}}{K_{m}} [\mbox{E}] [\mbox{S}]](images/math/3/0/8/3086604b8c68e57106b594af26800d13.png)
![\frac{1}{v} = \frac{K_{m}}{V_{max} [\mbox{S}]} + \frac{1}{V_{max}}](images/math/7/d/8/7d811128bb3a3302bed6b26a7215b5ce.png)
![\alpha = 1 + \frac{[\mbox{I}]}{K_{i}}](images/math/5/d/0/5d0ef5c67ccd4c852e6cfca33a00b3c1.png) y
y ![\alpha^{\prime} = 1 + \frac{[\mbox{I}]}{K_{i}^{\prime}}](images/math/1/3/1/131b809d1c25e2a337f97992bbbc9593.png)
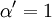 )
)
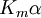
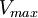
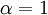 )
)
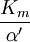
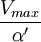
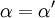 )
)
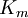
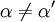 )
)
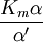

Acerca de quimica.es
Descubra toda la información interesante sobre nuestro portal especializado quimica.es.
Acerca de LUMITOS
Descubra más información sobre la empresa LUMITOS y nuestro equipo.
Anunciarse en LUMITOS
Descubra cómo puede ayudarle LUMITOS en su marketing online.
Los portales especializados de LUMITOS







© 1997-2024 LUMITOS AG, All rights reserved

Enciclopedia