|
Producto destacadoLas dos secuencias a alinear, llamadas A y B en los ejemplos, de longitud | A | = m y | B | = n, están formadas por elementos de un alfabeto finito de símbolos.
El algoritmo necesita saber qué símbolos son diferentes entre si y cuáles son iguales. Podemos utilizar una matriz cuadrada (S) para este propósito, en la que cada elemento Sij indique la similitud entre los elementos i y j del alfabeto usado. Si nuestro alfabeto de símbolos no fuese finito, en vez de una matriz podríamos usar una función Por ejemplo podemos definir la siguiente matriz:
Y entonces el siguiente alineamiento: AGACTAGTTAC CGA---GACGT con una penalización por hueco de d = − 5 nos devolvería como solución óptima: Para determinar la puntuación óptima y poder reconstruir el alineamiento que devolvería esa puntuación se necesita otra matriz, F. esta matriz almacena los resultados parciales de cada posible alineamiento. Es una matriz cuyas dimensiones son el número de elementos en la secuencia A por el de B ( | A | x | B | ).
Inicio del algoritmo: F0j = d * j Fi0 = d * i Recursión para obtener el siguiente elemento de forma óptima: Fij = max(Fi − 1,j − 1 + S(Ai,Bj),Fi,j − 1 + d,Fi − 1,j + d) La matriz F se calcula con el siguiente algoritmo: for i=0 to length(A)-1
F(i,0) <- d*i
for j=0 to length(B)-1
F(0,j) <- d*j
for i=1 to length(A)
for j = 1 to length(B)
{
Choice1 <- F(i-1,j-1) + S(A(i), B(j))
Choice2 <- F(i-1, j) + d
Choice3 <- F(i, j-1) + d
F(i,j) <- max(Choice1, Choice2, Choice3)
}
Cuando el algoritmo acaba tenemos calculada la matriz F; el resultado es la puntuación devuelta por el mejor alineamiento posible, de acuerdo a los parámetros que hemos definido. Para obtener la secuencia se necesita ejecutar el siguiente algoritmo, que hace uso de la matriz F. este algoritmo comienza por el último elemento, Fmn y va retrocediendo hasta llegar a un elemento de la primera fila o la primera columna de F. En cada paso se comparan 3 elementos de F para ver cuál de ellos es el que se ha seguido en la solución óptima. Para cada Fij debemos comparar Fi − 1,j,Fi,j − 1 y Fi − 1,j − 1. Si el elemento usado es Fi − 1,j, entonces Ai se ha alineado con un hueco; si es Fi,j − 1, entonces Bi se ha alineado con un hueco; y si no, si el elemento elegido es Fi − 1,j − 1, los elementos Ai y Bi han sido alineados. Es importante destacar que el que dos elementos sean alineados no implica necesariamente que sean iguales; significa que entre esa posibilidad, alinear con huecos o alinear símbolos diferentes, esa era la mejor opción. El pseudo-algoritmo que permite obtener el alineamiento correcto es el siguiente: AlignmentA <- ""
AlignmentB <- ""
i <- length(A) - 1
j <- length(B) - 1
while (i > 0 AND j > 0)
{
Score <- F(i,j)
ScoreDiag <- F(i - 1, j - 1)
ScoreUp <- F(i, j - 1)
ScoreLeft <- F(i - 1, j)
if (Score == ScoreDiag + S(A(i), B(j)))
{
AlignmentA <- A(i-1) + AlignmentA
AlignmentB <- B(j-1) + AlignmentB
i <- i - 1
j <- j - 1
}
else if (Score == ScoreLeft + d)
{
AlignmentA <- A(i-1) + AlignmentA
AlignmentB <- "-" + AlignmentB
i <- i - 1
}
otherwise (Score == ScoreUp + d)
{
AlignmentA <- "-" + AlignmentA
AlignmentB <- B(j-1) + AlignmentB
j <- j - 1
}
}
while (i > 0)
{
AlignmentA <- A(i-1) + AlignmentA
AlignmentB <- "-" + AlignmentB
i <- i - 1
}
while (j > 0)
{
AlignmentA <- "-" + AlignmentA
AlignmentB <- B(j-1) + AlignmentB
j <- j - 1
}
Se puede demostrar formalmente que tanto el tiempo de ejecución como el espacio necesario para ejecutar el algoritmo son de orden O(nm). Para alguna aplicaciones, sobre todo en bioinformática, el requerimiento de espacio es prohibitivo, puesto que se alinean secuencias muy largas. existe una optimización de este algoritmo, denominado algoritmo de Hirschberg que sólo necesita espacio del orden O(m+n), pero a costa de incrementar el tiempo de computación. Sitios externos
Véase también
Referencias
Categoría: Bioinformática |
|||||||||||||||||||||||||
| Este articulo se basa en el articulo Algoritmo_Needleman-Wunsch publicado en la enciclopedia libre de Wikipedia. El contenido está disponible bajo los términos de la Licencia de GNU Free Documentation License. Véase también en Wikipedia para obtener una lista de autores. |



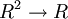 que tuviese como parámetros ambos símbolos a comparar y cuya salida fuese la similitud entre ambos. También se necesita otro parámetro (d) que nos indique cómo vamos a valorar que un símbolo no sea alineado con otro, y que en su lugar se utilice un hueco.
que tuviese como parámetros ambos símbolos a comparar y cuya salida fuese la similitud entre ambos. También se necesita otro parámetro (d) que nos indique cómo vamos a valorar que un símbolo no sea alineado con otro, y que en su lugar se utilice un hueco.
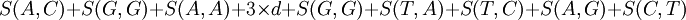

Acerca de quimica.es
Descubra toda la información interesante sobre nuestro portal especializado quimica.es.
Acerca de LUMITOS
Descubra más información sobre la empresa LUMITOS y nuestro equipo.
Anunciarse en LUMITOS
Descubra cómo puede ayudarle LUMITOS en su marketing online.
Los portales especializados de LUMITOS







© 1997-2026 LUMITOS AG, All rights reserved

