|
BLOSUM (BLOcks of Amino Acid SUbstitution Matrix, o matriz de sustitución de bloques de aminoácidos) es una matriz de sustitución utilizada para el alineamiento de secuencias de proteínas. BLOSUM se usa para puntuar alineamientos entre secuencias de proteínas evolutivamente divergentes. Se basa en alineamientos locales, y se introdujo en 1992 por primera vez en un paper de Henikoff y Henikoff[1] . Recorrieron la base de datos BLOCKS analizando regiones muy conservadas de familias de proteínas (sin huecos en el alineamiento de secuencias) y comprobaron las frecuencias relativas de aparición de los aminoácidos y las probabilidades de sustitución entre ellos. Seguidamente calcularon una puntuación de log-probabilidad para cada una de las 210 posibles sustituciones de los 20 aminoácidos estándar. Todas las BLOSUM se basan en alineamientos observados, y no son extrapoladas de comparaciones de proteínas cercanamente relacionadas (como es el caso de las matrices PAM, obtenidas al multiplicar por sí misma un determinado número de veces una matriz inicial). Producto destacadoExisten bastantes conjuntos de matrices BLOSUM que utilizan diferentes bases de datos de alineamientos, y que se nombran con números. Las BLOSUM seguidas de un número alto están diseñadas para comparar secuencias cercanamente relacionadas, mientras que las BLOSUM con número bajo están diseñadas para comparar secuencias relacionadas de forma distante. Por ejemplo, BLOSUM 80 se usa para alineamientos menos divergentes, mientras que BLOSUM 45 se usa para alineamientos más divergentes. Las puntuaciones dentro de una matriz BLOSUM corresponden a log-probabilidades que reflejan, en un alineamiento, el logaritmo de la razón de la probabilidad de la aparición de dos aminoácidos de una forma biológicamente intencionada o aceptada (residuos homológos; este numerador es la probabilidad de la hipótesis que queremos contrastar) y la probabilidad de su aparición por casualidad (el denominador es la probabilidad de la hipótesis nula).[2] Las matrices se basan en el mínimo porcentaje de identidad de la secuencia de proteína alineada usada al calcularlas (por ejemplo. BLOSUM 45 correspondería a alineamientos con un mínimo de un 45% de identidad).[3] A cada posible identidad o sustitución se le asigna una puntuación basada en las frecuencias observadas en el alineamiento de proteínas relacionadas.[4] Se da una puntuación positiva a las sustituciones más probables, mientras que corresponde una puntuación negativa para sustituciones menos probables. BLOSUM 62 es la matriz calculada usando las sustituciones observadas entre proteínas que tienen, como mínimo, el 62% de identidad en la secuencia, y se ha convertido en el estándar de la mayoría de los programas que utilizan este tipo de matrices.[2] BLOSUM ha demostrado actuar mejor en la puntuación de secuencias distantemente relacionadas que las otrora ampliamente usadas matrices Point Accepted Mutation (PAM).[5] Para calcular una matriz BLOSUM se usa la siguiente ecuación:
Aquí, pij es la probabilidad de que dos aminoácidos i y j reemplacen uno al otro en una secuencia homóloga, mientras que qi y qj son las probabilidades últimas de encontrar los aminoácidos i y j en cualquier secuencia de proteína de forma aleatoria. El factor λ es un mero factor de escala para asegurar que, tras su aplicación y la de un necesario redondeo al entero más cercano, la matriz contenga valores enteros dispersos y fácilmente tratables. De acuerdo a la definición vista, el logaritmo de la ecuación (el resultado de la ecuación, en definitiva) será positivo siempre que el cociente sea mayor de uno. Esto significará que la probabilidad de alineamiento entre los dos aminoácidos en una determinada secuencia se dará con mayor frecuencia que la que podríamos esperar por la mera casualidad. En resumen: esta sustitución es aceptada (en mayor o menor grado, de acuerdo a sus resultados estadísticos) por la evolución. Por el contrario, un logaritmo nulo o negativo implica que las sustituciones se dan al mismo (o menor) ritmo que las esperadas aleatoriamente. Referencias
Enlaces externos
Véase también |
| Este articulo se basa en el articulo BLOSUM publicado en la enciclopedia libre de Wikipedia. El contenido está disponible bajo los términos de la Licencia de GNU Free Documentation License. Véase también en Wikipedia para obtener una lista de autores. |



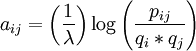

Acerca de quimica.es
Descubra toda la información interesante sobre nuestro portal especializado quimica.es.
Acerca de LUMITOS
Descubra más información sobre la empresa LUMITOS y nuestro equipo.
Anunciarse en LUMITOS
Descubra cómo puede ayudarle LUMITOS en su marketing online.
Los portales especializados de LUMITOS







© 1997-2026 LUMITOS AG, All rights reserved

