|
Si bien la mecánica cuántica es una teoría muy precisa para describir el comportamiento de los sistemas muy pequeños, su habilidad de predicción se ve limitada por el hecho de que sus ecuaciones son demasiado complejas de resolver numéricamente o menos aún analíticamente. La Teoría del Funcional de la Densidad (DFT en sus siglas en inglés) es una formulación alternativa de la mecánica cuántica en la que la cantidad central es la densidad y no la función de ondas de muchos cuerpos. La ventaja es que la densidad es un objeto mucho más simple que dicha función de ondas y por lo tanto más fácil de calcular. La desventaja es que no conocemos de manera exacta las ecuaciones de la teoría del funcional de la densidad y debemos aproximarlas. Su aplicación es principalmente a la descripción del comportamiento de los electrones tanto en química como en física de la materia condensada, donde el objeto central es la densidad electrónica. Aunque también ha sido aplicada a otras áreas como la física nuclear. Producto destacado
HistoriaLas primeras nociones de una teoría del funcional de la densidad fueron desarrolladas por Thomas y Fermi en los años 20. Ellos calcularon la energía de un átomo, representando su energía cinética como un funcional de su densidad electrónica, y combinando esto con las expresiones clásicas de las interacciones núcleo-electrón y electrón-electrón (que también se pueden representar en términos de densidad electrónica). El modelo fue mejorado por Dirac, quien añadió un funcional de energía de canje en 1928. Sin embargo, la teoría de Thomas-Fermi-Dirac era imprecisa para la mayoría de las aplicaciones, por la mala representación de la energía cinética con un funcional de densidad. La base teórica para la DFT fue dada en 1964 por Hohenberg y Kohn, quienes mostraron que la energía es un funcional de la densidad y que además la densidad del sistema minimiza este funcional. Sin embargo, el desarrollo más importante fue dado el año siguiente, cuando Kohn y Sham demostraron que a partir de la teoría del funcional de la densidad es posible escribir una ecuación para orbitales de una partícula, de los cuales se obtiene la densidad. La DFT era muy popular para cálculos de física del estado sólido desde los '70. Sin embargo, se consideraba que no era lo bastante precisa para la química cuántica hasta los '90, cuando se refinaron en gran medida las aproximaciones usadas en la teoría. Ahora la DFT es un método fundamental para los cálculos de estructura electrónica en ambos campos. Descripción de la teoría
Un funcional es una función que recibe como argumento otra función. En mecánica cuántica estándar el objeto básico es la función de onda y la energía es un funcional de dicha función de onda. A cualquier función de onda podemos asignarle una energía y además para la función de onda física, la energía será mínima. En la teoría del funcional de la densidad, el objeto básico es la densidad y la energía es un funcional de ésta. Además, se cumple un principio variacional: la energía será mínima para la densidad real del sistema. Se ha demostrado matemáticamente que existe una relación uno a uno entre la densidad y la función de onda, por lo que en principio se puede utilizar indistintamente una u otra. Desde el punto de vista numérico, la función de onda es un objeto muy complejo de manipular, pues para N partículas es una función de 3N variables, mientras que la densidad es más fácil de manejar pues es siempre una función de 3 variables, independientemente del número de partículas. La desventaja es que si bien DFT es una teoría exacta, no conocemos de forma explícita las ecuaciones exactas que debe cumplir la densidad por lo que sólo podemos aproximarlas. En la mecánica cuántica estándar, los observables son calculados a partir de la función de onda de muchos cuerpos. El método de la DFT fue sometido a un tratamiento riguroso por Hohenberg y Kohn [1] en 1964, quienes demostraron que todo observable (propiedad) de un sistema electrónico puede ser calculado a partir de la densidad electrónica. Esto quiere decir que la densidad contiene en principio la misma información que la función de onda. En particular, Hohenberg y Kohn mostraron que la energía es un funcional de la densidad y que además la densidad del sistema minimiza este funcional. El problema, es que no no se conoce la forma completa de este funcional, por lo que debe ser aproximado. Kohn y Sham presentaron una forma de aproximar el energía cinética en 1965 [2], mapeando el problema de la densidad a un sistema de ecuaciones monoelectrónicas. Para esto se incorpora el concepto de orbital de métodos de función de ondas, aunque estos orbitales y sus autoenergías no tienen significación física directa. Intercambio y correlaciónAún con esta aproximación, no se conoce la forma funcional para las energías de intercambio (también llamado canje) y correlación electrónicas. Estas corresponden a la interacción cuántica entre electrones, la primera debido al principio de exclusión de Pauli entre electrones del mismo espín y el segundo debido a la parte cuántica de la repulsión coulombiana. Aproximación de Densidad LocalLa primera aproximación para este funcional se conoce como Aproximación de Densidad Local (LDA) y consiste en suponer que en cada punto, la energía de intercambio y correlación depende sólo de la densidad en ese punto. Este valor se considera como el que tendría un gas de electrones libres de esa densidad. Si bien es una aproximación bastante fuerte, se obtienen resultados sorprendentemente precisos para algunas propiedades, y es en parte a eso que se debe el éxito de esta teoría. Aproximaciones de Gradiente GeneralizadoExisten aproximaciones más sofisticadas para el funcional de intercambio y correlación, estas se conocen como Aproximaciones de Gradiente Generalizado , estas son semilocales, ya que consideran en cada punto el valor de la densidad y sus gradientes. Para algunas propiedades estas aproximaciones dan mejores resultados que LDA, en particular para geometrías moleculares y energías del estado fundamental, aunque para otras no representan una mejora sustancial. Funcionales dependientes de los orbitales e Intercambio exacto (EXX)Una serie de funcionales más sofisticados puede obtenerse al suponer que la energía de intercambio y correlación depende explícitamente de los orbitales de Kohn-Sham. El más común de estos funcionales es el de Intercambio Exacto (EXX), que incluye de manera completa la energía de intercambio electrónico y que puede derivarse desde primeros principios. El problema de este tipo de funcionales es que computacionalmente son más costosos de tratar. ¿Ab initio o no?Existe una controversia sobre si la Teoría del Funcional de la Densidad puede ser considerada o no un método ab initio. En general en Física se le considera así, debido a que no se requiere ningún tipo de parámetro adicional ni ajuste obtenido de resultados experimentales. En Química por el contrario, suele guardarse el termino ab initio para métodos derivados de la Teoría cuántica de muchos cuerpos que por lo general son más precisos, más costosos computacionalmente y cuyo nivel de aproximación puede ser ajustado. AplicacionesLa principal ventaja de la Teoría del Funcional de la Densidad es que las ecuaciones de esta son mucho más simples de resolver que las ecuaciones de muchos cuerpos de mecánica cuántica u otras aproximaciones, por lo que permiten tratar sistemas más grandes y calcular mas propiedades. Por lo general es posible llegar a hacer simulaciones con unos pocos miles de átomos. El principal problema es que si bien es en principio una teoría exacta, solo se puede aplicar de forma aproximada, lo que hace que sus resultados sean menos precisos que otros métodos. Además, diferentes aproximaciones a la energía de intercambio y correlación pueden dar resultados diferentes. Aun así, para muchos métodos más sofisticados se utiliza como punto de partida los resultados de DFT. En 1998 Walter Kohn recibió el premio Nobel de Química por sus aportes al desarrollo de esta teoría. CódigosExisten varios programas (o códigos) computacionales que utilizan la teoría del funcional de la densidad para calcular propiedades de sistemas electrónicos como moléculas, sólidos o cúmulos. Un Ejemplo de cálculos de primeros principios utilizando la teoría del funcional de la DensidadUtilizando la teoría del funcional de la densidad, es posible calcular las propiedades estructurales de sistemas fisicos. En particular la teoría es útil para describir dichas propiedades en los materiales semiconductores. Este ejemplo muestra resultados sobre la estructura más estable del Silicio y el tipo de enlace que presenta en la estructura de tipo diamante.
En el caso la estructura tipo diamante se obtuvo la energía y la densidad de carga, en las otras tres solamente se obtuvo la energía. Obtención de la Energía en función del Volumen de la celda primitiva.Para cada estructura se realizarán ciclos autoconsistentes que resuelven las ecuaciones de Khon-Sham para diferentes volúmenes. A partir de los resultados se obtiene la energía de cohesión, Que es la resta de la energía total para la estructura menos la energía atómica. Los datos obtenidos para las diferentes estructuras fueron ajustados a la ecuación de Murnaghan para la energía de cohesión, obteniéndose los valores del módulo de bulto y su derivada. La ecuación de Murnaghan para la energía es la siguiente:
Y los resultados de los cálculos realizados se resumen en la tabla siguiente:
El valor experimental para el parámetro de red de la estructura diamante es de 5.43 [A]4. Al comparar los resultados obtenidos encontramos que la estructura más estable es la de tipo diamante, esta se encuentra -1.214 [eV] por debajo de la estructura cúbica simple. Con lo que se comprueba que la estructura mas etable es la de tipo diamante. Obtención de la Densidad de Carga y Densidad de Estados. El tipo de enlace y la naturaleza química de los semiconductores es un punto de interés en la física del estado sólido. El énfasis en el tipo de enlace es motivado por la idea de que al tener una descripción del enlace uno puede tener mayor información de la distribución de carga electrónica dentro de los sólidos lo cual nos conduciría a un mejor entendimiento de las propiedades físicas y químicas de los objetos en estudio. Esta información se puede obtener también utilizando calculos de primeros principios. Para poder obtener la información deseada proponemos que nuestro sistema de estudio sea el de los cristales covalentes, los atomos de silicio se localizarían en las posiciones de los nodos de la red cristalina. La característica principal de un cristal covalente es que los atomos dentro de este se encuentran ligados por medio de un enlace covalente. El enlace más fuerte ocurre cuando los espines de los electrones en el dentreo de el son antiparalelos. El enlace depende de la orientación relativa de los espines no por que exista una interacción de fuerza magnética fuerte debida a los dipolos magnéticos formados, sino por que el principio de exclusión de Pauli modifica la distribución de carga de acuerdo a la orientación de espines. Esta dependencia de espines de la energía coulombiana es llamada la interacción de intercambio. El principio de exclusión de Pauli genera una interacción de tipo repulsiva fuerte entre los átomos y las capas ocupadas por los electrones. Si las capas no se encuentran ocupadas, el traslape de los electrones puede ser reacomodado sin tomar en cuenta a los electrones excitados y el enlace seria más corto. Cálculos de primeros principios para la densidad de carga son utilizados ampliamente para poder obtener información sobre la distribución de electrones dentro de los cristales al mismo tiempo que se utiliza para calcular las cargas de enlace covalente. La distribución espacial de la densidad de carga para una banda de valencia n puede ser escrita como
Donde la suma corre sobre todos los estados permitidos i en la banda n. Los resultados de los cálculos son detallados en las superficies de nivel de la densidad de carga de valencia total. La grafica se realizó con la paquetería contenida en espresso-2.1.4 con el programa PWscF. La densidad de carga es graficada en curvas de nivel para el elemento en el plano cristalino (110).
Las esferas en negro representan las posiciones de los núcleos de los átomos de silicio y las regiones sombreadas representan la densidad de carga de los electrones. En la grafica se observa claramente que en la región intersticial entre los átomos existe un enlace de tipo covalente, donde la densidad de carga se encuentra concentrada justo por la mitad entre los iones.
La grafica anterior muestra la densidad de carga como función de x donde la variable x va de una posición del Si dentro del cristal a otra posición del silicio. La densidad de estados (DOS) es una propiedad utilizada comúnmente en física de materia que cuantifica que tan estrechamente se encuentran los niveles de energía empaquetados en algún sistema físico. Es a menudo expresada como una función de la energía interna. Esto por lo general es usado con niveles de energía electrónicos en un sólido. Para este trabajo se calculó la densidad de estados para la estructura del diamante y los resultados se muestran en la gráfica siguiente.
El nivel de Fermi para el silicio esta marcado con la línea este tiene un valor de 5.48 [eV]. Los estados a la izquierda de ella representan los estados ocupados y los de la derecha representan a los desocupados. Mediante la´presentación de estas graficas se concluye el ejemplo aquí desarrollado, estas nos dan una muestra visual de la estructura más estable, de el tipo de enlace y la densidad de estados del silicio en la estructura tipo diamante. Todas estas propiedades fueron obtenidos con calculos de primeros principios dentro del formalismo de la teoría del funcional de la densidad.
Comparación con otros métodosHay cierta división en la comunidad científica. Los defensores de la DFT indican que sus resultados son muy satisfactorios, y que, por su bajo coste computacional, es la única forma de abordar sistemas más allá de cierta complejidad. Sus detractores apuntan a que es un método semiempírico más, y que no es tan fiable como los métodos ab initio "clásicos". ExtensionesLa teoría del funcional de la densidad ha sido generalizada para tratar sistemas más complejos. Una de las generalizaciones más importantes es la Teoría del funcional de la densidad tiempo-dependiente (TDDFT por sus siglas en inglés) que permite ampliar la teoría para el estudio de sistemas bajo excitaciones.
Software de ayuda para DFT
Referencias[1] P. Hohenberg y W. Kohn, Phys. Rev. 136 (1964) B864 LiteraturaKlaus Capelle, Una visión a vista de pájaro de la teoría del funcional de densidad. Categoría: Química cuántica |
|
| Este articulo se basa en el articulo Teoría_del_funcional_de_la_densidad publicado en la enciclopedia libre de Wikipedia. El contenido está disponible bajo los términos de la Licencia de GNU Free Documentation License. Véase también en Wikipedia para obtener una lista de autores. |


![E\left( V\right)=E_{0}+\frac{B_{0}V}{B_{0}^{\prime }}% \left[ \frac{\left( \frac{V_{0}}{V}\right) ^{B_{0}^{\prime }}}{B_{0}^{\prime }-1}+1\right] -\frac{B_{o}V_{0}}{B_{0}^{\prime}-1}](images/math/6/1/3/6139f8916548d487ccbe15aaf7f10efa.png)
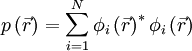

Acerca de quimica.es
Descubra toda la información interesante sobre nuestro portal especializado quimica.es.
Acerca de LUMITOS
Descubra más información sobre la empresa LUMITOS y nuestro equipo.
Anunciarse en LUMITOS
Descubra cómo puede ayudarle LUMITOS en su marketing online.
Los portales especializados de LUMITOS







© 1997-2026 LUMITOS AG, All rights reserved

