La técnica de aprendizaje de la máquina acelera la determinación de la estructura de los cristales
Anuncios



Los nanoingenieros de la Universidad de California en San Diego han desarrollado un método computarizado que podría hacer menos intensiva la labor de determinar las estructuras cristalinas de varios materiales y moléculas, incluyendo aleaciones, proteínas y productos farmacéuticos. El método utiliza un algoritmo de aprendizaje automático, similar al utilizado en el reconocimiento facial y en los coches que se conducen solos, para analizar independientemente los patrones de difracción de electrones, y hacerlo con al menos un 95% de precisión.
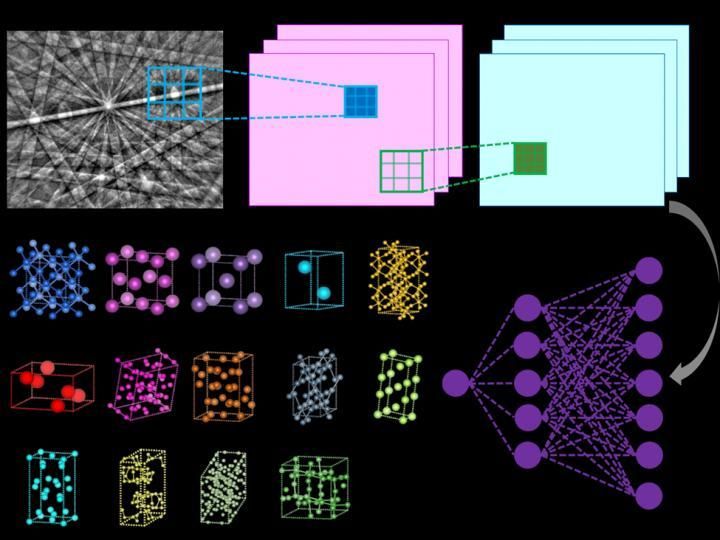
Ilustración del funcionamiento interno de una red neural convolutiva que calcula la probabilidad de que el patrón de difracción de entrada pertenezca a una clase determinada (por ejemplo, la red de Bravais o el grupo espacial).
Vecchio lab/Science
Un equipo dirigido por el profesor de nanoingeniería de la Universidad de California en San Diego, Kenneth Vecchio, y su estudiante de doctorado Kevin Kaufmann, que es el primer autor del artículo, desarrollaron el nuevo enfoque. Su método implica el uso de un microscopio electrónico de barrido (SEM) para recoger patrones de difracción de retrodispersión de electrones (EBSD). En comparación con otras técnicas de difracción de electrones, como las de la microscopía electrónica de transmisión (MET), la EBSD basada en la MET puede realizarse en muestras grandes y analizarse a múltiples escalas de longitud. Esto proporciona información local submicrónica mapeada a escalas de centímetros. Por ejemplo, un sistema moderno de EBSD permite determinar las estructuras de grano a escala fina, las orientaciones de los cristales, la tensión o tensión residual relativa y otra información en un solo escaneo de la muestra.
Sin embargo, el inconveniente de los sistemas comerciales de EBSD es la incapacidad del software para determinar la estructura atómica de las redes cristalinas presentes en el material analizado. Esto significa que un usuario del software comercial debe seleccionar hasta cinco estructuras cristalinas que se presume que están en la muestra y luego el software intenta encontrar coincidencias probables con el patrón de difracción. La compleja naturaleza del patrón de difracción hace que el software encuentre a menudo falsas coincidencias de estructura en la lista seleccionada por el usuario. Por consiguiente, la precisión de la determinación del tipo de red por parte del programa informático existente depende de la experiencia y el conocimiento previo del operador de su muestra.
El método que el equipo de Vecchio desarrolló hace todo esto de forma autónoma, ya que la red neural profunda analiza independientemente cada patrón de difracción para determinar la red cristalina, de todos los tipos de estructura de red posibles, con un alto grado de precisión (más del 95%).
Se espera que una amplia gama de áreas de investigación, incluyendo la farmacología, la biología estructural y la geología se beneficien del uso de algoritmos automatizados similares para reducir la cantidad de tiempo necesario para la identificación estructural de los cristales, dijeron los investigadores.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original
"Crystal symmetry determination in electron diffraction using machine learning"; Science; 2020




Más noticias del departamento ciencias