El aprendizaje por máquina rompe el enigma de la química cuántica
Las técnicas de inteligencia artificial calculan con precisión la energía necesaria para hacer - o romper - moléculas simples
Anuncios



Una nueva herramienta de aprendizaje de la máquina puede calcular la energía necesaria para hacer - o romper - una molécula con mayor precisión que los métodos convencionales. Mientras que la herramienta actualmente sólo puede manejar moléculas simples, prepara el camino para futuros conocimientos en química cuántica.
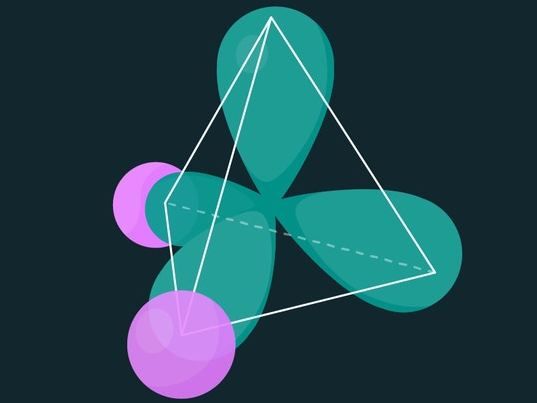
La distribución electrónica tetraédrica de una molécula de agua. El núcleo del átomo de oxígeno está en el centro del tetraedro, y los núcleos de hidrógeno están en el centro de las esferas rosadas.
Simons Foundation
"El uso del aprendizaje automático para resolver las ecuaciones fundamentales que rigen la química cuántica ha sido un problema abierto durante varios años, y hay mucho entusiasmo en torno a él en este momento", dice el co-creador Giuseppe Carleo, un científico investigador del Centro de Física Cuántica Computacional del Instituto Flatiron en la ciudad de Nueva York. Una mejor comprensión de la formación y destrucción de las moléculas, dice, podría revelar el funcionamiento interno de las reacciones químicas vitales para la vida.
Carleo y sus colaboradores Kenny Choo de la Universidad de Zurich y Antonio Mezzacapo del IBM Thomas J. Watson Research Center en Yorktown Heights, Nueva York, presentaron su trabajo el 12 de mayo en Nature Communications.

La herramienta del equipo estima la cantidad de energía necesaria para ensamblar o separar una molécula, como el agua o el amoníaco. Ese cálculo requiere determinar la estructura electrónica de la molécula, que consiste en el comportamiento colectivo de los electrones que unen la molécula.
La estructura electrónica de una molécula es algo difícil de calcular, ya que requiere la determinación de todos los estados potenciales en los que podrían estar los electrones de la molécula, más la probabilidad de cada estado.
Dado que los electrones interactúan y se enredan mecánicamente cuánticos entre sí, los científicos no pueden tratarlos individualmente. Con más electrones, surgen más enredos y el problema se vuelve exponencialmente más difícil. No existen soluciones exactas para moléculas más complejas que los dos electrones que se encuentran en un par de átomos de hidrógeno. Incluso las aproximaciones luchan con la exactitud cuando involucran más que unos pocos electrones.
Uno de los retos es que la estructura electrónica de una molécula incluye estados para un número infinito de orbitales que se alejan cada vez más de los átomos. Además, un electrón es indistinguible de otro, y dos electrones no pueden ocupar el mismo estado. Esta última regla es una consecuencia de la simetría de intercambio, que rige lo que sucede cuando partículas idénticas cambian de estado.
Mezzacapo y sus colegas de IBM Quantum desarrollaron un método para limitar el número de orbitales considerados e imponer la simetría de intercambio. Este enfoque, basado en los métodos desarrollados para las aplicaciones de la informática cuántica, hace que el problema se asemeje más a los escenarios en los que los electrones están confinados a lugares preestablecidos, como en una red rígida.
La similitud con las celosías rígidas fue la clave para hacer el problema más manejable. Carleo previamente entrenó redes neuronales para reconstruir el comportamiento de los electrones confinados a los sitios de una red. Al extender esos métodos, los investigadores pudieron estimar soluciones a los problemas de compactación de Mezzacapo. La red neuronal del equipo calcula la probabilidad de cada estado. Usando esta probabilidad, los investigadores pueden estimar la energía de un estado dado. El nivel de energía más bajo, llamado energía de equilibrio, es donde la molécula es más estable.
Las innovaciones del equipo hicieron que el cálculo de la estructura electrónica de una molécula básica fuera más simple y rápido. Los investigadores demostraron la exactitud de sus métodos al estimar cuánta energía se necesitaría para separar una molécula del mundo real, rompiendo sus enlaces. Hicieron cálculos para el dihidrógeno (H2), el hidruro de litio (LiH), el amoníaco (NH3), el agua (H2O), el carbono diatómico (C2) y el dinitrógeno (N2). Para todas las moléculas, las estimaciones del equipo demostraron ser muy precisas incluso en rangos donde los métodos existentes tienen dificultades.
En el futuro, los investigadores pretenden abordar moléculas más grandes y complejas utilizando redes neuronales más sofisticadas. Uno de los objetivos es manejar productos químicos como los que se encuentran en el ciclo del nitrógeno, en el que los procesos biológicos construyen y rompen las moléculas basadas en el nitrógeno para hacerlas utilizables para la vida. "Queremos que esta sea una herramienta que pueda ser utilizada por los químicos para procesar estos problemas", dice Carleo.
Carleo, Choo y Mezzacapo no son los únicos que están aprendiendo a resolver problemas en la química cuántica. Los investigadores presentaron su trabajo por primera vez en arXiv.org en septiembre de 2019. En ese mismo mes, un grupo en Alemania y otro en el DeepMind de Google en Londres, cada uno de ellos publicó una investigación utilizando el aprendizaje automático para reconstruir la estructura electrónica de las moléculas.
Los otros dos grupos usan un enfoque similar entre sí que no limita el número de orbitales considerados. Esta inclusión, sin embargo, es más gravosa desde el punto de vista computacional, un inconveniente que sólo empeorará con moléculas más complejas. Con los mismos recursos computacionales, el enfoque de Carleo, Choo y Mezzacapo produce una mayor precisión, pero las simplificaciones realizadas para obtener esta precisión podrían introducir sesgos.
"En general, es una compensación entre el sesgo y la precisión, y no está claro cuál de los dos enfoques tiene más potencial para el futuro", dice Carleo. "Sólo el tiempo nos dirá cuál de estos enfoques se puede ampliar a los desafiantes problemas abiertos de la química".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original




Más noticias del departamento ciencias