Resultados muy precisos a bajo coste computacional
Los investigadores logran predecir los parámetros de RMN con la aproximación DLPNO
Anuncios



La resonancia magnética nuclear (RMN) es una de las técnicas analíticas más importantes utilizadas en la investigación química, farmacéutica y biomédica, así como en las ciencias de los materiales. Proporciona información detallada sobre las estructuras de las moléculas en disolución, que es, sin embargo, indirecta. De ahí que los investigadores tengan que recurrir a menudo a herramientas de la química teórica para interpretar adecuadamente los complejos datos experimentales. Una de las principales fuentes de información en los espectros de RMN es el llamado "desplazamiento químico" que existe para cada átomo de una molécula y que depende de su entorno. Los químicos cuánticos simulan el desplazamiento químico para cada átomo, con el fin de recibir todo el contenido de información de los espectros de RMN. Sin embargo, los métodos utilizados hasta ahora han alcanzado repetidamente sus límites.
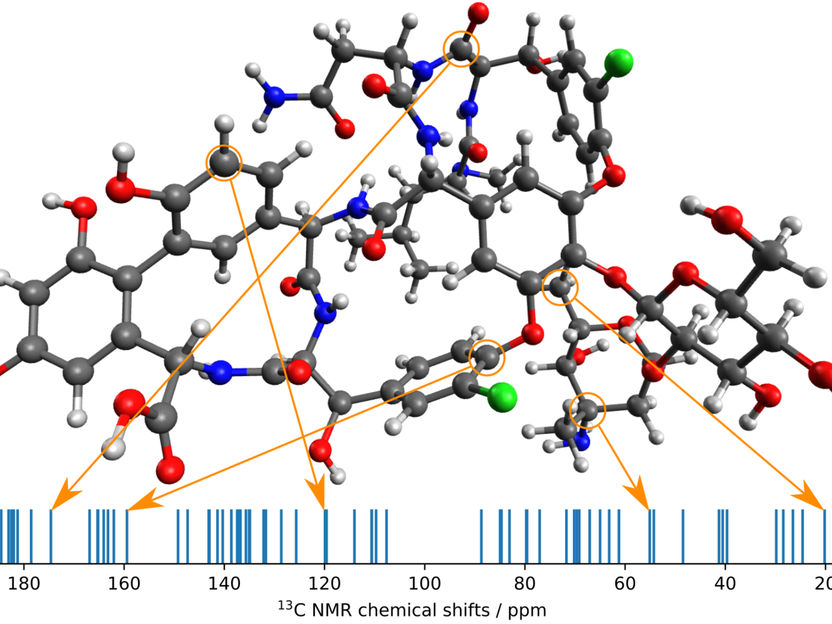
El nuevo método de los investigadores del MPI en aplicación. Un conformador de la molécula de vancomicina y su espectro de RMN de 13C calculado con algunos picos asignados a átomos.
© MPI für Kohlenforschung
Los resultados anteriores solían ser demasiado imprecisos o requerían un gran esfuerzo computacional; la nueva aproximación DLPNO reduce considerablemente el esfuerzo computacional de los métodos exactos
La combinación de experimentos y nuevos métodos teóricos conduce a conocimientos que no habrían sido accesibles por otros medios. Sin embargo, los científicos se enfrentan a menudo a un dilema: métodos eficientes como la teoría del funcionamiento de la densidad -el caballo de batalla de la química teórica- suelen ofrecer una precisión insuficiente. Por su parte, los métodos de alta precisión de la función de onda requieren muchos más recursos computacionales y su ámbito de aplicación se limita a moléculas con pocos átomos, demasiado pequeñas para aplicaciones exitosas en la investigación farmacéutica o biomédica.
Investigadores del Departamento de Teoría Molecular y Espectroscopia del MPI für Kohlenforschung han desarrollado ahora un nuevo enfoque que supone un gran avance. Su método de función de onda se basa en el llamado marco de orbitales naturales de pares locales basados en dominios (DLPNO), un concepto desarrollado por Frank Neese. En un artículo publicado recientemente, Stoychev et al. informan de cómo han conseguido calcular con precisión los desplazamientos químicos de sistemas con cientos de átomos. Su nuevo método ahorra tiempo de cálculo y abre interesantes perspectivas para los usuarios, ya que en muchas áreas de la química, la interpretación de los espectros de RMN forma parte del trabajo diario.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original



Más noticias del departamento ciencias

































