Un nuevo método mejora la precisión de los potenciales aprendidos por máquina para simular catalizadores
Un puente entre precisión y eficacia en el diseño de catalizadores
Anuncios



Los catalizadores desempeñan un papel indispensable en la fabricación moderna. Más del 80% de todos los productos manufacturados, desde los farmacéuticos hasta los plásticos, dependen de procesos catalíticos en alguna fase de la producción. Los metales de transición, en particular, destacan como catalizadores muy eficaces porque sus orbitales d parcialmente llenos les permiten intercambiar electrones fácilmente con otras moléculas. Sin embargo, esta misma propiedad dificulta su modelización, que requiere descripciones precisas de su estructura electrónica.
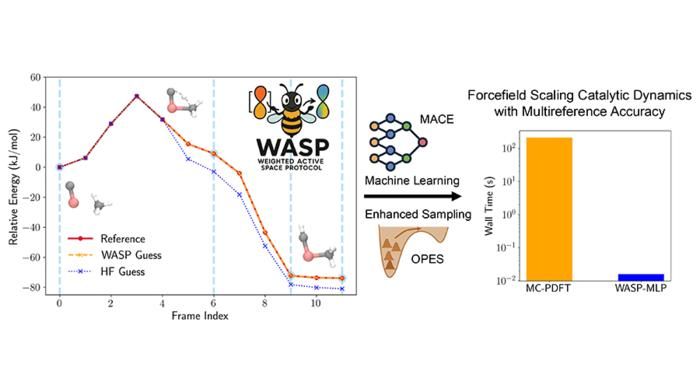
Precisión y aceleración logradas con el Protocolo de Espacio Activo Ponderado (WASP) para la activación de metano en carburo de titanio.
Figure courtesy of Seal et al.
Diseñar catalizadores eficientes de metales de transición que puedan funcionar en condiciones realistas requiere algo más que una instantánea estática de una reacción. Hay que captar la imagen dinámica, es decir, cómo se mueven e interactúan las moléculas a diferentes temperaturas y presiones, donde el movimiento atómico determina fundamentalmente el rendimiento catalítico.
Para hacer frente a este reto, el laboratorio de la profesora Laura Gagliardi, de la Escuela Pritzker de Ingeniería Molecular de la Universidad de Chicago (UChicago PME) y del Departamento de Química, ha desarrollado una nueva y potente herramienta que aprovecha las teorías de la estructura electrónica y el aprendizaje automático para simular la dinámica catalítica de los metales de transición con precisión y rapidez.

"A lo largo de la última década, los potenciales aprendidos por máquinas han hecho avanzar significativamente la forma en que simulamos la dinámica molecular, ofreciendo velocidad y escalabilidad. Sin embargo, capturar con precisión la estructura electrónica de los catalizadores de metales de transición sigue siendo un reto sin resolver. Nuestro nuevo método salva esta laguna integrando métodos de química cuántica multirreferencia con potenciales aprendidos por máquina, ofreciendo precisión y eficiencia". afirma Gagliardi.
Los resultados se publicaron en Proceedings of the National Academy of Sciences.
Aprendizaje automático para acelerar las simulaciones
Durante la última década, el grupo de Gagliardi ha desarrollado la teoría del funcional de densidad de pares multiconfiguración (MC-PDFT), un método de química cuántica capaz de describir las intrincadas estructuras electrónicas de las reacciones de los metales de transición. Aunque la MC-PDFT ofrece una gran precisión, es prohibitivamente lenta para simular la dinámica de los sistemas catalíticos, un paso fundamental para predecir cómo se comportan realmente los catalizadores en condiciones realistas.
Para hacer frente a este reto, el equipo recurrió a los potenciales interatómicos aprendidos por máquina (potenciales ML), que pueden capturar la dinámica molecular con notable eficacia. Los potenciales ML se han aplicado ampliamente en la ciencia de materiales, pero hasta ahora nunca se habían combinado con éxito con métodos multirreferencia como MC-PDFT.
La razón radica en un antiguo obstáculo: la consistencia del etiquetado. Los modelos de aprendizaje automático requieren etiquetas de propiedades únicas y fiables -como energías y fuerzas derivadas de funciones de onda- para cada geometría molecular a lo largo de una ruta de reacción. En el caso de los métodos de química cuántica multirreferencia, la asignación inequívoca de estas etiquetas seguía siendo un problema sin resolver.
Para superar este reto, el estudiante de doctorado Aniruddha Seal, asesorado conjuntamente por Gagliardi y el profesor Andrew Ferguson, desarrolló un novedoso algoritmo que genera funciones de onda coherentes para nuevas geometrías como una combinación ponderada de funciones de onda de estructuras moleculares previamente muestreadas. Cuanto más se parece una geometría nueva a otra conocida, más se asemeja su función de onda a la de la estructura conocida. Este enfoque garantiza que a cada punto a lo largo de una ruta de reacción se le asigne una función de onda única y coherente, lo que hace posible entrenar potenciales ML con precisión en datos multirreferencia.
"Es como mezclar pinturas en una paleta", explica Seal. "Si quiero crear un tono de verde más cercano al azul, utilizaré más pintura azul y sólo un poco de amarilla. Si quiero un tono que se incline hacia el amarillo, la balanza se invierte. Cuanto más se acerque mi color objetivo a una de las pinturas base, más influirá en la mezcla. WASP funciona de la misma manera: mezcla información de estructuras moleculares cercanas, dando más peso a las que son más similares, para crear una predicción precisa de la nueva geometría".
Esta innovación constituye la base del Protocolo de Espacio Activo Ponderado (WASP), un marco que combina la precisión de MC-PDFT con la eficacia del aprendizaje automático, desarrollado gracias a una estrecha colaboración con el Grupo Parrinello del Instituto Italiano de Tecnología de Génova, que aúna experiencia en teoría de estructuras electrónicas y potenciales aprendidos por máquinas. WASP ofrece una velocidad espectacular: simulaciones con precisión multirreferencia que antes tardaban meses ahora pueden completarse en cuestión de minutos.
Impacto: Unir precisión y eficiencia en el diseño de catalizadores
Al aunar precisión y velocidad, WASP abre la puerta al diseño de catalizadores capaces de soportar condiciones realistas: altas temperaturas y altas presiones. Los metales de transición son fundamentales en innumerables procesos a gran escala, pero su complejidad ha dificultado el diseño racional de catalizadores.
Un buen ejemplo es el proceso Haber-Bosch, en el que el hierro sirve de catalizador para convertir el nitrógeno y el hidrógeno en amoníaco. A pesar de haberse desarrollado hace más de un siglo, este catalizador de hierro sigue dominando la producción de amoníaco en todo el mundo. Con WASP, los investigadores disponen ahora de herramientas para explorar alternativas que podrían aumentar la eficiencia, reducir los subproductos y disminuir el coste medioambiental.
Hasta ahora, WASP ha demostrado su eficacia en la catálisis activada térmicamente, es decir, en las reacciones impulsadas por el calor. La próxima frontera es adaptar el método a las reacciones activadas por la luz, esenciales para el diseño de nuevos fotocatalizadores. Los fotocatalizadores son muy prometedores para tecnologías que van desde el tratamiento del agua hasta la producción de energía.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original


Más noticias del departamento ciencias










































