Una herramienta eficaz para vincular los experimentos con rayos X y la teoría ab initio
Los resultados pueden incluso calcularse durante el experimento
Anuncios



La estructura electrónica de las moléculas complejas y su reactividad química pueden evaluarse mediante el método de dispersión inelástica de rayos X (RIXS) en BESSY II. Sin embargo, la evaluación de los datos RIXS ha requerido hasta ahora tiempos de computación muy largos. Un equipo de BESSY II ha desarrollado ahora un nuevo método de simulación que acelera enormemente esta evaluación. Los resultados pueden calcularse incluso durante el experimento. Los usuarios invitados podrían utilizar el procedimiento como una caja negra.
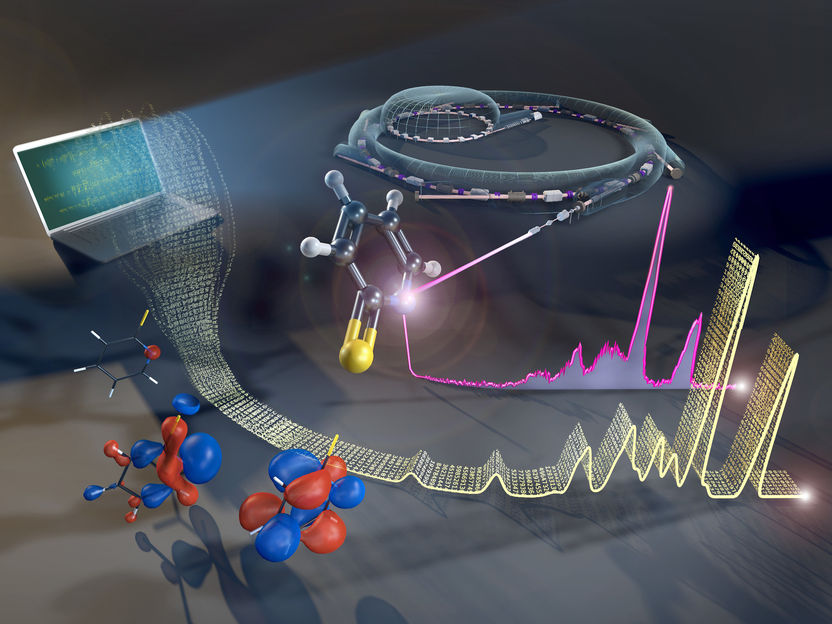
La estructura electrónica de las moléculas complejas puede evaluarse mediante el método de dispersión inelástica de rayos X (RIXS) en BESSY II.
© Martin Künsting /HZB
Las moléculas formadas por muchos átomos son estructuras complejas. Los electrones exteriores se distribuyen entre los distintos orbitales, y su forma y ocupación determinan el comportamiento químico y la reactividad de la molécula. La configuración de estos orbitales puede analizarse experimentalmente. Las fuentes de sincrotrón, como BESSY II, ofrecen un método para ello: La dispersión inelástica de rayos X resonante (RIXS). Sin embargo, para obtener información sobre los orbitales a partir de los datos experimentales, es necesario realizar simulaciones químicas cuánticas. Los tiempos de cálculo típicos para las moléculas más grandes tardan semanas, incluso en ordenadores de alto rendimiento.
Acelerar la evaluación
"Hasta ahora, estos cálculos se han realizado en su mayoría después de las mediciones", explica el químico teórico Dr. Vinicius Vaz da Cruz, postdoc en el equipo del Prof. Dr. Alexander Föhlisch. Junto con el experto en RIXS, el Dr. Sebastian Eckert, también postdoctoral en el equipo de Föhlisch, han desarrollado un nuevo y sofisticado procedimiento que acelera mucho la evaluación.
"Con nuestro método, se tarda unos minutos y no necesitamos un superordenador para ello, sino que funciona en máquinas de sobremesa", dice Eckert. Los científicos del HZB han probado el método con la molécula 2-tiopiridona, un sistema modelo para la transferencia de protones, que son procesos esenciales en las células y organismos vivos. A pesar del escaso tiempo de cálculo, los resultados son lo suficientemente precisos como para ser muy útiles.
"Es un gran paso adelante", subraya Föhlisch. "Podemos recorrer muchas opciones de antemano y llegar a conocer la molécula, por así decirlo. Además, este método también permite simular moléculas mucho más complejas e interpretar de forma significativa los datos obtenidos experimentalmente". El físico experimental Eckert añade: "Ahora también podemos ejecutar las simulaciones durante la medición y ver inmediatamente dónde puede ser especialmente interesante echar un vistazo más de cerca".
El procedimiento es una extensión de la teoría funcional de la densidad dependiente del tiempo, bien establecida y muy eficiente, que es mucho más rápida que los conceptos tradicionales para simular el proceso RIXS. "La simplicidad del método permite un gran grado de automatización", afirma Vaz da Cruz: "Puede utilizarse como una caja negra".
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original




Más noticias del departamento ciencias