El aprendizaje automático acelera las simulaciones en la ciencia de los materiales
Los métodos más rápidos y precisos resultan útiles en diversas aplicaciones, desde el almacenamiento de energía hasta los medicamentos
Anuncios



La investigación, el desarrollo y la producción de nuevos materiales dependen en gran medida de la disponibilidad de métodos de simulación rápidos y al mismo tiempo precisos. El aprendizaje automático, en el que la inteligencia artificial (IA) adquiere y aplica de forma autónoma nuevos conocimientos, pronto permitirá a los investigadores desarrollar sistemas materiales complejos en un entorno puramente virtual. ¿Cómo funciona esto y qué aplicaciones se beneficiarán? En un artículo publicado en la revista Nature Materials, un investigador del Instituto Tecnológico de Karlsruhe (KIT) y sus colegas de Gotinga y Toronto lo explican todo.
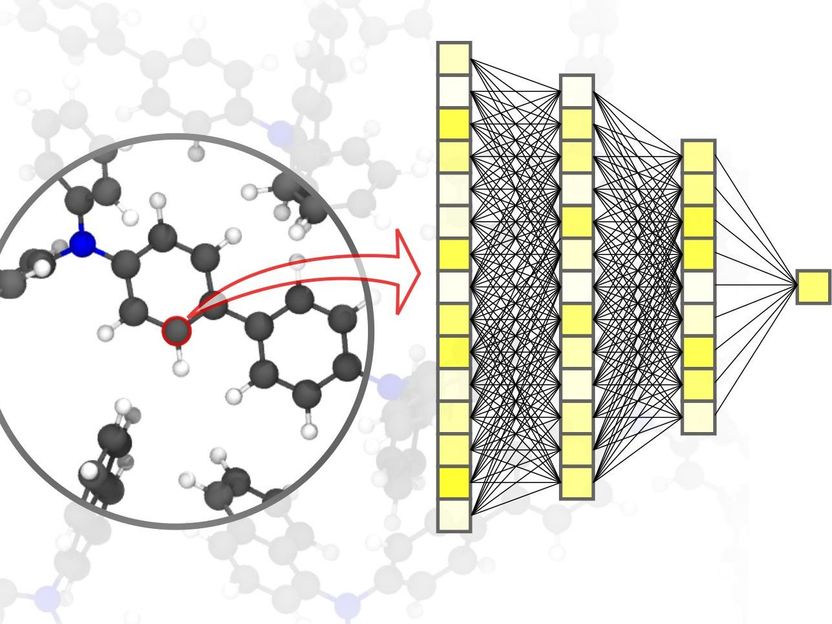
Las redes neuronales permiten realizar simulaciones precisas en la ciencia de los materiales, hasta el nivel de los átomos individuales.
Pascal Friederich, KIT
La digitalización y la virtualización son cada vez más importantes en una amplia gama de disciplinas científicas. Una de ellas es la ciencia de los materiales: la investigación, el desarrollo y la producción de nuevos materiales dependen en gran medida de la disponibilidad de métodos de simulación rápidos y, al mismo tiempo, precisos. Esto, a su vez, es beneficioso para un amplio abanico de aplicaciones diferentes: desde los sistemas de almacenamiento de energía eficientes, como los indispensables para el uso de energías renovables, hasta los nuevos medicamentos, para cuyo desarrollo se requiere la comprensión de complejos procesos biológicos. La IA y los métodos de aprendizaje automático pueden llevar las simulaciones en las ciencias de los materiales a un nivel superior. "En comparación con los métodos de simulación convencionales basados en cálculos clásicos o de mecánica cuántica, el uso de redes neuronales específicamente adaptadas a las simulaciones de materiales nos permite lograr una importante ventaja de velocidad", explica el físico y experto en IA, el profesor Pascal Friederich, director del grupo de investigación AiMat - Inteligencia Artificial para las Ciencias de los Materiales, del Instituto de Informática Teórica (ITI) del KIT. "Con sistemas de simulación más rápidos, los científicos podrán desarrollar sistemas de materiales más grandes y complejos en un entorno puramente virtual, y comprenderlos y optimizarlos hasta el nivel atómico."
Alta precisión desde el átomo hasta el material
En un artículo publicado en Nature Materials, Pascal Friederich, que también es jefe de grupo asociado de la división de Nanomateriales por Diseño Guiado por la Información del Instituto de Nanotecnología (INT) del KIT, presenta, junto con investigadores de la Universidad de Göttingen y la Universidad de Toronto, una visión general de los principios básicos del aprendizaje automático utilizado para las simulaciones en las ciencias de los materiales. Esto incluye también el proceso de adquisición de datos y los métodos de aprendizaje activo. Los algoritmos de aprendizaje automático no sólo permiten a la inteligencia artificial procesar los datos de entrada, sino también encontrar patrones y correlaciones en grandes conjuntos de datos, aprender de ellos y hacer predicciones y decisiones autónomas. Para las simulaciones en la ciencia de los materiales, es importante lograr una gran precisión en diferentes escalas de tiempo y tamaño, desde el átomo hasta el material, al tiempo que se limitan los costes computacionales. En su artículo, los científicos también hablan de varias aplicaciones actuales, como las pequeñas moléculas orgánicas y las grandes biomoléculas, los materiales sólidos, líquidos y gaseosos estructuralmente desordenados, así como los sistemas cristalinos complejos - por ejemplo, los marcos metal-orgánicos que pueden utilizarse para el almacenamiento de gases o para la separación, para los sensores o para los catalizadores.
Más velocidad con los métodos híbridos
Para ampliar aún más las posibilidades de las simulaciones de materiales en el futuro, los investigadores de Karlsruhe, Göttingen y Toronto sugieren el desarrollo de métodos híbridos: éstos combinan métodos de aprendizaje automático (ML) y de mecánica molecular (MM). Las simulaciones de MM utilizan los llamados campos de fuerza para calcular las fuerzas que actúan sobre cada partícula individual y predecir así los movimientos. Como los potenciales de los métodos ML y MM son bastante similares, es posible una estrecha integración con zonas de transición variables. Estos métodos híbridos podrían acelerar significativamente la simulación de grandes biomoléculas o reacciones enzimáticas en el futuro, por ejemplo.
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.
Publicación original



Más noticias del departamento ciencias






























