El poder de las salamanquesas
La Universidad Técnica de Viena resuelve el rompecabezas de las grandes moléculas
Anuncios

La Universidad Técnica de Viena ha resuelto un enigma de la química teórica: un nuevo método computacional permite calcular las fuerzas entre moléculas de gran tamaño con una precisión sin precedentes.
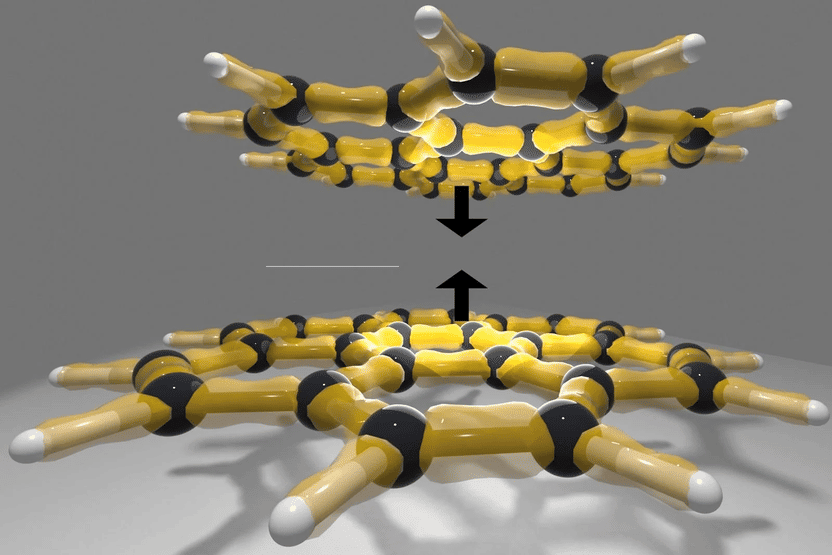
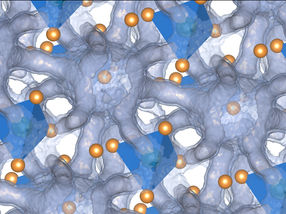
Coroneno-dímero: un ejemplo de moléculas grandes con fuerzas de Van-der-Waals
© TU Wien
¿Por qué las salamanquesas pueden trepar por las paredes? ¿Por qué el nitrógeno se vuelve líquido a -196 °C? Muchos fenómenos cotidianos se explican por las fuerzas de van der Waals, enlaces débiles entre moléculas muy difíciles de calcular. Durante años, los científicos se han enfrentado al hecho de que distintos métodos computacionales arrojaban resultados contradictorios.
Ahora, investigadores de la Universidad Técnica de Viena han resuelto esta discrepancia y han encontrado una solución. Irónicamente, fue el propio método considerado durante mucho tiempo el "patrón oro" de la química cuántica el que resultó ser la fuente del error: sobrestima sistemáticamente la energía contenida en ciertos enlaces moleculares. Con una variante mejorada, el equipo de la TU Wien puede ahora predecir correctamente el comportamiento de moléculas de gran tamaño, un paso esencial para comprender los sistemas biológicos y avanzar en las tecnologías de energías renovables.

Un misterio de la química
"Para describir los enlaces entre moléculas grandes, los científicos utilizan distintos enfoques computacionales", explican Tobias Schäfer y Andreas Irmler, primeros autores del nuevo estudio. Junto con Alejandro Gallo y el jefe del grupo de investigación, el profesor Andreas Grüneis, compararon los métodos más utilizados.
"Una opción es utilizar simulaciones cuánticas de Montecarlo", explica Schäfer. "Aquí, el ordenador explora innumerables disposiciones posibles de electrones, manteniendo las energéticamente favorables y descartando las desfavorables". Otra opción es el llamado enfoque de clúster acoplado", añade Irmler. "En ese caso, las moléculas se tratan en sus estados de baja energía, y las configuraciones de mayor energía se añaden después como una especie de corrección".
"Este método de clústeres acoplados se ha considerado durante mucho tiempo el método de referencia", afirma Schäfer. "Pero cuanto más lo analizábamos, más claro se hacía que había pequeñas pero persistentes desviaciones en comparación con los resultados de Monte Carlo, y durante años nadie supo por qué".
Ahora, el equipo de TU Wien ha encontrado la respuesta: "Descubrimos que el método de clústeres acoplados sobrestima sistemáticamente las energías de enlace en moléculas grandes y muy polarizables", explica Irmler. "Nuestra variante mejorada corrige esta desviación sin aumentar significativamente el coste computacional". Con esta corrección, los resultados se ajustan ahora mucho más a los datos de Monte Carlo cuántico.
Moléculas grandes, gran importancia
Este avance es especialmente crucial para los grandes sistemas moleculares. "Si queremos describir moléculas que contienen hasta cien átomos, el esfuerzo computacional es enorme", explica Alejandro Gallo. "Incluso los mayores superordenadores del mundo alcanzan sus límites. Para lograr predicciones fiables, necesitamos métodos de aproximación muy sofisticados".
Y las moléculas grandes son cada vez más importantes, en campos que van desde la investigación de materiales al desarrollo farmacéutico. "Si queremos entender cómo cristaliza un fármaco dentro de una pastilla, o con qué fuerza se une un material al hidrógeno para almacenar energía, necesitamos modelizar con precisión las fuerzas de van der Waals", dice Schäfer.
De la teoría fundamental a las aplicaciones prácticas
El nuevo método permite obtener datos de referencia más fiables, no sólo para las simulaciones tradicionales, sino también como datos de entrenamiento para modelos de inteligencia artificial. Estos modelos ya se utilizan para diseñar nuevos materiales y productos farmacéuticos en entornos virtuales.
"Estamos construyendo un puente entre la máxima precisión y la utilidad práctica", afirma el profesor Andreas Grüneis, del Instituto de Física Teórica de la Universidad Técnica de Viena. "Esto abre nuevas posibilidades para la ciencia de los materiales. Nuestros resultados demuestran que incluso los métodos bien establecidos deben reexaminarse continuamente para seguir el ritmo de las crecientes exigencias de la investigación moderna."
Nota: Este artículo ha sido traducido utilizando un sistema informático sin intervención humana. LUMITOS ofrece estas traducciones automáticas para presentar una gama más amplia de noticias de actualidad. Como este artículo ha sido traducido con traducción automática, es posible que contenga errores de vocabulario, sintaxis o gramática. El artículo original en Inglés se puede encontrar aquí.

Más noticias del departamento ciencias